Test Files
Click here for an example bed file using the Saccharomyces cerevisiae genome [1] or here for an example fasta file from Human Creb[2].
1. Lefrancois, P., Euskirchen, G. M., Auerbach, R. K., Rozowsky, J., Gibson, T., Yellman, C. M., . . . Snyder, M. (2009). Efficient yeast ChIP-Seq using multiplex short-read DNA sequencing. BMC Genomics, 10, 37. doi:10.1186/1471-2164-10-37
2. Zhang, X., Odom, D. T., Koo, S. H., Conkright, M. D., Canettieri, G., Best, J., . . . Montminy, M. (2005). Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A, 102(12), 4459-4464. doi:10.1073/pnas.0501076102
Usage
Trawler is a motif discovery tool designed with biologists in mind. Simply input your sample sequences as bed format and select your organism. The organism names are based on the nomenclature used by UCSC.
Organims provided by Trawler:
| Organism | Accession Number |
|---|---|
| Zebrafish | danRer7 |
| Medaka | oryLat2 |
| Stickleback | gasAcu1 |
| Human | hg19 |
| Mouse | mm9 |
| mm10 | |
| Rat | rn5 |
| Marmoset | calJac3 |
| Chicken | galGal3 |
| Xenopus tropicalis | xenTro3 |
| Fruit fly | dm3 |
| Caenorhabditis elegans | ce10 |
| Saccharomyces cerevisiae | sacCer3 |
| Arabidopsis thaliana | TAIR10 |
The sample data may be uploaded as a .txt or .bed file or alternatively pasted directly into the textbox. When uploading data as bed format, ensure that the columns are tab separated with the first column containing the chromosome name eg: "chr1" and the second and third columns containing the chromosome start and end respectively.
Sample BED format:chr1 123456 894834
chr2 123456 899012
chrY 123456 688929
If using FASTA format, a background dataset must be provided. This background dataset should be representative of the sample distribution relative to the distance from the regulating gene or nearest gene. All FASTA sequences should be repeat-masked with 'N'.
Additional options
The options provided are set to default parameters and can be changed accordingly. The options currently available are: motif database, strand, motif length, percentage overlap, occurrence, wildcard, number of motifs and number of clusters.
The motif database contains a list of all known TFBS from JASPAR and UniPROBE. This list is used to compare known TFs with the indentified motif(s). The default database is set to vertebrates but can be changed to either fungi, plants, insects, nematodes or all depending on the source of the input sample. e.g: selecting "insects" when analysing ChIP-seq data from drosophila.
The strand option indicates whether Trawler will search through both + and - strands of DNA for the motif or just the + strand. By default, Trawler will search both strands (double option).
The minimum motif length sets the minimum length of motifs discovered. Values accepted are restricted between 6 to 20 inclusive.
Percentage overlap is the percentage of similarity required between two instances to be clustered within the same cluster.
The occurrence gives the number of times a motif appears in the sample sequence. The lower limit is defined by 2. However, 10 to 20 is generally a good minimum occurrence.
The number of wildcards gives the maximum amount of positions in the motif with degenerate nucleotides.
The number of motifs sets the maximum number of motifs to be used for clustering. If more than the set amount is identified, the best number of motifs are used.
By default Trawler will cluster motifs by SCC (strongly connected components) clustering if the number of clusters is set to zero. By setting the expected number of clusters, Trawler will cluster the motifs by the k-means clustering algorithm.
Output results
The main results tab displays a list of identified binding motifs in order of decreasing Z-score with an annotation of known TFBSs. For each motif family there is a separate tab containing a table displaying every instance of the motif identified in the sample. The motif instance can be displayed in UCSC by clicking on the chromosome region. This option is only available from bed inputted user data.
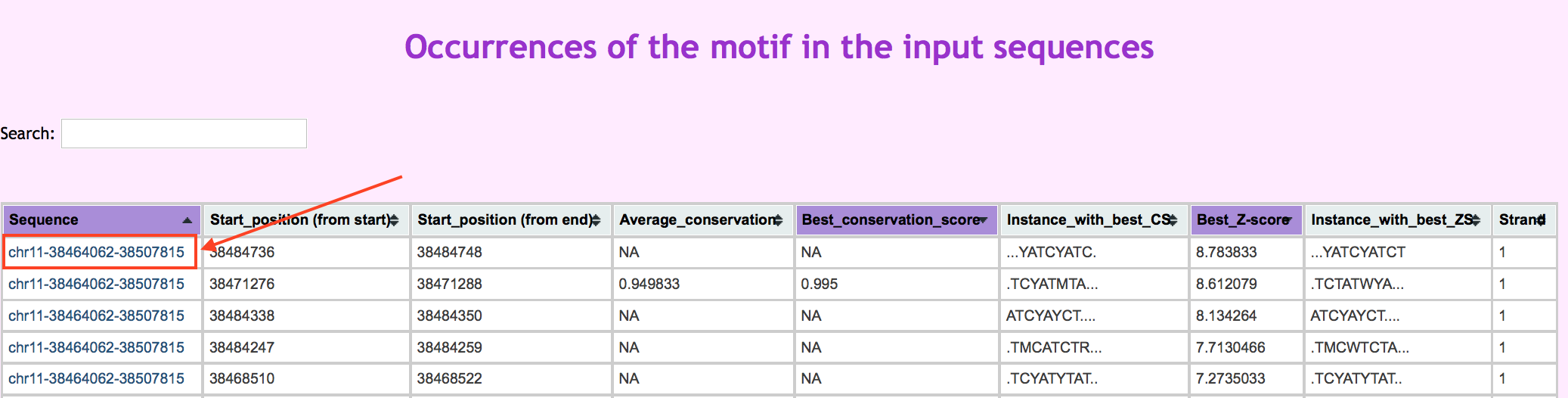
The first three columns in the table above shows a where the instance is found within the sample file. The "average consevation score" for each instance is calculated by taking the average score for the given instance using PhastCons scores. The "best conservation score" is taken as the base with the maximum PhastCons score within the sequence.
Please click here for an example output.
FAQs
Q: What is Trawler?
A: Trawler is a motif discovery tool used to identify enriched motifs in a set of sequenced regions of DNA.
Q: I have over 9000 regions, can I still use Trawler to find my motifs?
A: YES! Trawler has been used to run histone datasets upto 40,000 regions in total. However, Trawler is currently limited to sample files 26 megabases in total and a background file of 134 megabases in total.
Q: Trawler is telling me I don't have enough background. What should I do?
A: Trawler requires that the number of regions in the background is at least 5 times more than the number of sample regions. Additionally, make sure the size of the background regions are also larger than the sample regions.
Q: The organism I am working is not on the list, how can I analyse the dataset?
A: For different assemblies of the same organism, consider using the genome liftover tool provided by UCSC. Alternatively, you can run the data under FASTA format. If you think that others will benefit from having a certain genome available, shoot us an email and we can make it available provided the genome is well sequenced and annotated.
Q: Everytime I run Trawler with BED input, the results are slightly different with sometimes more or less motifs. Why does this happen and how can I get reproducible results?
A: The way Trawler is programmed, each time you input BED formatted sequences, it randomly generates a background for analysis. This randomness is responsible for differing results. For reproducible results and to account for this randomness, consider downloading both the background and sample files as FASTA format from the "downloads" tab and rerun the sequences under FASTA format.
Contacts:
Mirana Ramialison: mirana.ramialison@monash.edu
Louis Dang: louis.dang@monash.edu
Source code
The source code is available on gitHub.